
Principal Investigator
Mathieu Bertrand
Research interests
Our research group investigates how cell survival, regulated cell death, and inflammatory responses are controlled downstream of members of the TNF receptor superfamily and pattern recognition receptors. A defining aspect of our work is the systematic dissection of molecular checkpoints that protect cells from immune receptor–induced cytotoxicity, ensuring that inflammatory signalling does not inadvertently trigger cell death. These checkpoints function as critical control nodes that safeguard tissue integrity during immune responses.
Our research uncovers the pathological consequences of chronic inactivation of these safeguards, demonstrating how their sustained loss leads to excessive cell death and drives inflammatory disease. Recently, we have revealed the role of uncoventional forms of autophagy in restraining cell death and inflammatory signalling.
Importantly, our findings also show that these protective mechanisms are not merely passive safety brakes, but are integral components of host immune defence strategies against pathogens. When microbes attempt to suppress or hijack inflammatory signalling to promote their own survival, the controlled and context-dependent disabling of these checkpoints can deliberately shift the balance toward cell death. In this setting, transient and tightly regulated cell death acts as an immune “fail-safe”, eliminating infected cells and restricting pathogen replication.
Together, our work helps to reveal that cell death is not simply a detrimental by-product of inflammation, but rather a strategic backup mechanism that ensures immune effectiveness when conventional inflammatory pathways are compromised. This framework reshapes how inflammation, cell survival, and cell death are understood – not as opposing outcomes, but as coordinated and adaptable components of host defence.
- Cell Death
- Innate immunity
- Cell Death Checkpoints
- Autophagy
- Signal transduction
- Post-translational modifications
Selected publications
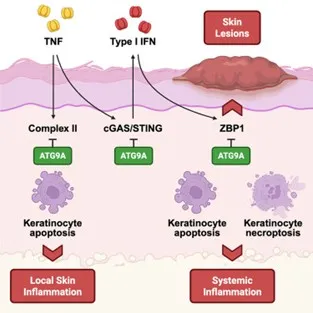
- Priem D, Huyghe J, Gilbert B, Verdonck S, Delanghe T, Verstraeten B, Hoste E, Vandenabeele P, Maelfait J, van Loo G, Bertrand MJM. ATG9A-mediated autophagy prevents inflammatory skin disease by limiting TNFR1-driven STING activation and ZBP1-dependent cell death. Immunity. 2025 Dec 9;58(12):2972-2988.e6.
- Atg9aΔKer mice develop a TNF-dependent cell death-driven inflammatory skin disorder
- ATG9A inhibits TNFR1 complex II-driven apoptosis to prevent local skin inflammation
- ATG9A restrains TNFR1-mediated cGAS/STING activation and subsequent ZBP1 expression
- ATG9A represses ZBP1 cytotoxicity to prevent skin lesions and systemic inflammation
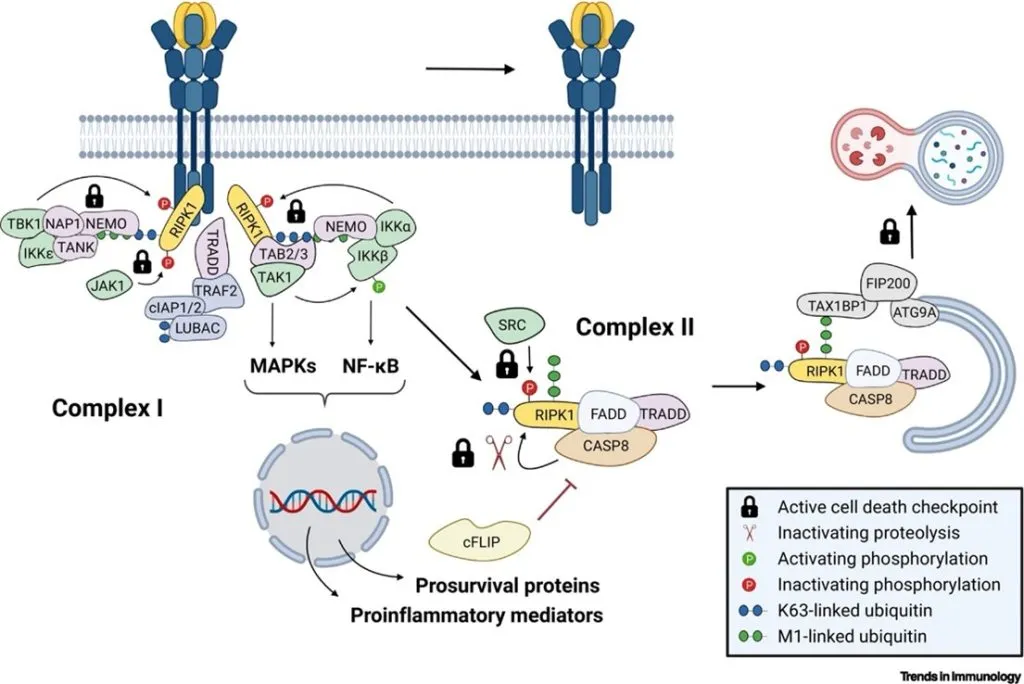
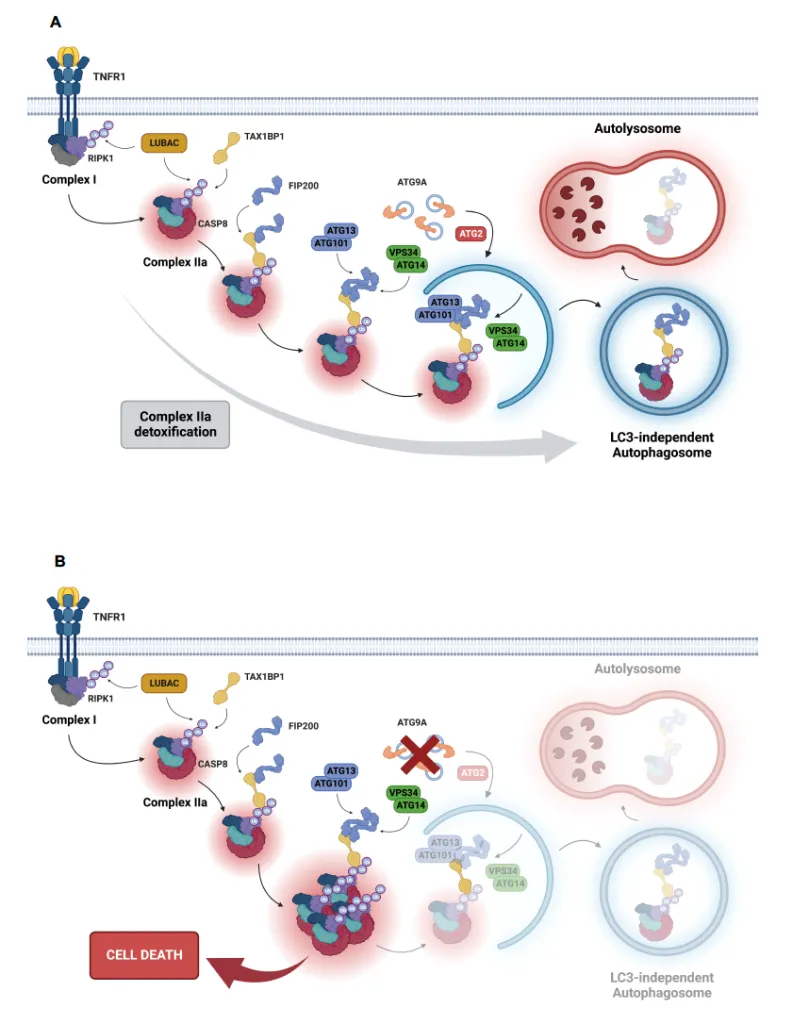
- Huyghe J, Priem D, Van Hove L, Gilbert B, Fritsch J, Uchiyama Y, Hoste E, van Loo G, Bertrand MJM. ATG9A prevents TNF cytotoxicity by an unconventional lysosomal targeting pathway. Science 2022 Dec 16;378(6625):1201-1207.
- ATG9A and FIP200 are key components of a new checkpoint that protects cells from TNF-induced, RIPK1 kinase–independent apoptosis
- The checkpoint operates through an unconventional LC3-independent autophagy pathway
- The autophagy receptor TAX1BP1 recognizes M1-ubiquitinated RIPK1 within complex IIa and recruits FIP200 to initiate its encapsulation and degradation, bypassing the need for LC3
- Inhibiting the checkpoint, through ATG9A loss, leads to TNFR1-dependent embryonic lethality due to excessive liver apoptosis and causes TNF-driven inflammatory skin disease when deleted in keratinocytes
- Delanghe T, Vadi M, Haems A, Wijns L, Bruggeman I, Huyghe J, Priem D, Vandenabeele P, Bertrand MJ. TAB2 controls a TAK1-independent cell death checkpoint at the level of TNFR1 complex II in the TNF pathway. Cell Death Differ. 2026 Jan;33(1):64-76.
- TAB2 deficiency switches the TNF response from survival to apoptosis
- TAK1 remains functional in absence of TAB2
- TAB2 is an integral part of TNFR1 complex II, limiting the abundance of the cytotoxic complex through direct association
- The ubiquitin-binding NZF domain of TAB2 is critical for its TAK1-independent pro-survival function
- Dondelinger Y, Delanghe T, Priem D, Wynosky-Dolfi MA, Sorobetea D, Rojas-Rivera D, Giansanti P, Roelandt R, Gropengiesser J, Ruckdeschel K, Savvides SN, Heck AJR, Vandenabeele P, Brodsky IE, Bertrand MJM. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat Commun. 2019 Apr 15;10(1):1729.
- Phosphorylation of RIPK1 on Ser25 by IKKs directly inhibits RIPK1 kinase activity and prevents TNF-mediated RIPK1 kinase-dependent cell death
- Mimicking Ser25 phosphorylation (S > D mutation) protects cells and mice from the cytotoxic effect of TNF in conditions of IKK inhibition
- Mimicking Ser25 phosphorylation compromises the in vivo cell death-dependent immune control of Yersinia infection, a physiological model of TAK1/IKK inhibition
- Mimicking Ser25 phosphorylation rescues the cell death-induced multi-organ inflammatory phenotype of the SHARPIN-deficient mice.
